 中文网站
中文网站
Tidlig om morgenen 29. desember publiserte NEJM en ny klinisk fase III-studie av det nye kinesiske koronaviruset VV116 på nett. Resultatene viste at VV116 ikke var verre enn Paxlovid (nematovir/ritonavir) når det gjaldt varighet av klinisk bedring og hadde færre bivirkninger.

Bildekilde: NEJM
Median restitusjonstid 4 dager, bivirkningsrate 67,4 %
VV116 er et oralt nukleosid-anti-nytt koronavirus (SARS-CoV-2)-legemiddel utviklet i samarbeid med Junsit og Wang Shan Wang Shui, og er en RdRp-hemmer sammen med Gileads remdesivir, Merck Sharp & Dohmes molnupiravir og Real Biologics' azelvudin.
I 2021 ble en klinisk fase II-studie av VV116 fullført i Usbekistan. Resultatene av studien viste at VV116-gruppen bedre kunne forbedre kliniske symptomer og redusere risikoen for progresjon til kritisk form og død betydelig sammenlignet med kontrollgruppen. Basert på de positive resultatene fra denne studien er VV116 godkjent i Usbekistan for behandling av pasienter med moderat til alvorlig COVID-19, og har blitt det første nye orale koronarmedisinet som er godkjent for markedsføring utenlands i Kina [1].
Denne kliniske fase III-studien[2] (NCT05341609), ledet av professor Zhao Ren fra Shanghai Ruijin Hospital, professor Gaoyuan fra Shanghai Renji Hospital og akademiker Ning Guang fra Shanghai Ruijin Hospital, ble fullført under utbruddet forårsaket av Omicron-varianten (B.1.1.529) fra mars til mai i Shanghai, med mål om å evaluere effekten og sikkerheten til VV116 versus Paxlovid for tidlig behandling av pasienter med mild til moderat COVID-19. Målet var å evaluere effekten og sikkerheten til VV116 versus Paxlovid for tidlig behandling av pasienter med mild til moderat COVID-19.

Bildekilde: Referanse 2
En multisenter, observatørblindet, randomisert, kontrollert studie av 822 voksne covid-19-pasienter med høy risiko for progresjon og med milde til moderate symptomer ble gjennomført mellom 4. april og 2. mai 2022 for å vurdere kvalifiseringen til deltakere fra syv sykehus i Shanghai, Kina. Til slutt fikk 771 deltakere enten VV116 (384, 600 mg hver 12. time på dag 1 og 300 mg hver 12. time på dag 2-5) eller Paxovid (387, 300 mg nimatuvir + 100 mg ritonavir hver 12. time i 5 dager) som oral medisin.
Resultatene fra denne kliniske studien viste at tidlig behandling med VV116 for mild til moderat COVID-19 oppnådde det primære endepunktet (tid til vedvarende klinisk bedring) forutsagt av den kliniske protokollen: median tid til klinisk bedring var 4 dager i VV116-gruppen og 5 dager i Paxlovid-gruppen (risikoforhold, 1,17; 95 % KI, 1,02 til 1,36; nedre grense >0,8).
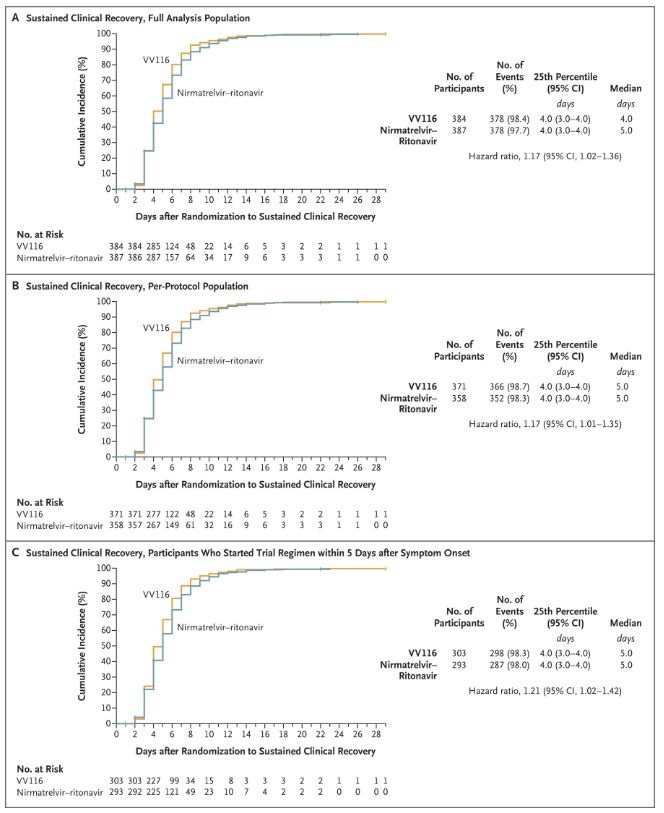
Opprettholdelse av klinisk restitusjonstid
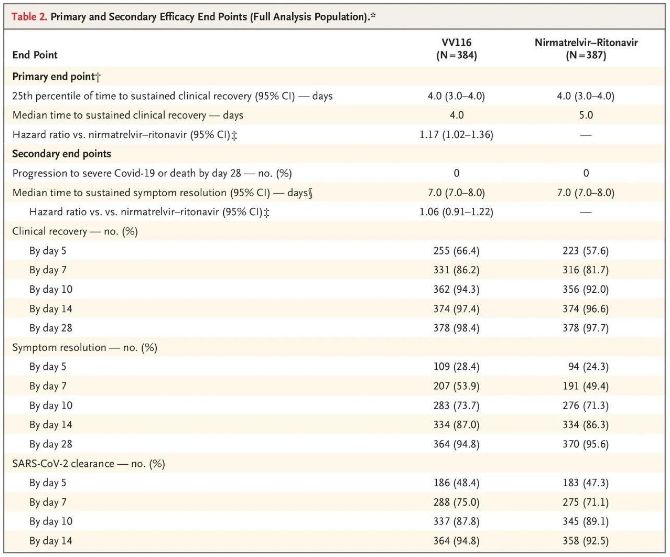
Primære og sekundære effektmål (omfattende analyse av populasjonen)
Bildekilde: Referanse 2
Når det gjelder sikkerhet, rapporterte deltakerne som fikk VV116 færre bivirkninger (67,4 %) enn de som fikk Paxlovid (77,3 %) ved 28-dagers oppfølging, og forekomsten av grad 3/4 bivirkninger var lavere for VV116 (2,6 %) enn for Paxlovid (5,7 %).
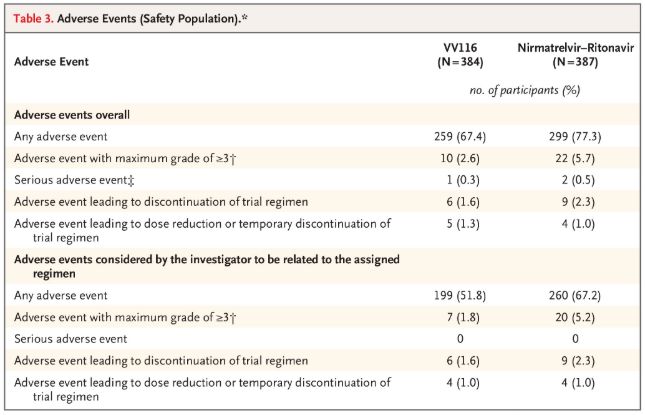
Uønskede hendelser (trygge personer)
Bildekilde: Referanse 2
Kontroverser og spørsmål
23. mai 2022 offentliggjorde Juniper at den kliniske fase III-registreringsstudien av VV116 versus PAXLOVID for tidlig behandling av mild til moderat COVID-19 (NCT05341609) nådde sitt primære endepunkt.

Bildekilde: Referanse 1
I en tid da detaljer om studien manglet, var kontroversen rundt fase III-studien todelt: for det første var det en enkeltblind studie, og i mangel av en placebokontroll fryktet man at det ville være vanskelig å bedømme legemidlet helt objektivt; for det andre var det spørsmål om de kliniske endepunktene.
De kliniske inklusjonskriteriene for Juniper er (i) positive resultater for den nye kronetesten, (ii) ett eller flere milde eller moderate COVID-19-symptomer, og (iii) pasienter med høy risiko for alvorlig COVID-19, inkludert død. Det eneste primære kliniske endepunktet er imidlertid «tid til vedvarende klinisk bedring».
Rett før kunngjøringen, 14. mai, hadde Juniper revidert de kliniske endepunktene ved å fjerne et av de kliniske primære endepunktene, «andel av konverteringer til alvorlig sykdom eller død» [3].
![]()
Bildekilde: Referanse 1
Disse to hovedstridspunktene ble også spesifikt behandlet i den publiserte studien.
På grunn av det plutselige utbruddet av Omicron var ikke produksjonen av placebotabletter for Paxlovid fullført før studiestart, og derfor kunne ikke forskerne gjennomføre denne studien med et dobbeltblindet, dobbelt-mock-design. Når det gjelder det enkeltblinde aspektet av den kliniske studien, sa Juniper at protokollen ble utført etter kommunikasjon med regulatoriske myndigheter, og at det enkeltblinde designet betyr at verken forsker (inkludert evaluatoren av studiens endepunkt) eller sponsoren vil vite den spesifikke terapeutiske legemiddelallokeringen før den endelige databasen er låst ved studiens slutt.
Frem til tidspunktet for den endelige analysen hadde ingen av deltakerne i studien opplevd død eller progresjon til en alvorlig Covid-19-hendelse, så det kan ikke trekkes noen konklusjoner om effekten av VV116 i å forhindre progresjon til alvorlig eller kritisk Covid-19 eller død. Dataene indikerte at den estimerte mediantiden fra randomisering til vedvarende regresjon av Covid-19-relaterte målsymptomer var 7 dager (95 % KI, 7 til 8) i begge gruppene (hazard ratio, 1,06; 95 % KI, 0,91 til 1,22) [2]. Det er ikke vanskelig å forklare hvorfor det primære endepunktet «konverteringsrate til alvorlig sykdom eller død», som opprinnelig ble satt før studiens slutt, ble fjernet.
Den 18. mai 2022 publiserte tidsskriftet Emerging Microbes & Infections resultatene av den første kliniske studien av VV116 hos pasienter infisert med Omicron-varianten [4], en åpen, prospektiv kohortstudie med 136 bekreftede innlagte pasienter.
Data fra studien viste at pasienter med Omicron-infeksjon som brukte VV116 innen 5 dager etter sin første positive nukleinsyretest, hadde en tid til nukleinsyreregresjon på 8,56 dager, mindre enn 11,13 dager i kontrollgruppen. Administrasjon av VV116 til symptomatiske pasienter innenfor tidsrammen for denne studien (2–10 dager etter første positive nukleinsyretest) reduserte tiden til nukleinsyreregresjon hos alle pasienter. Når det gjelder legemiddelsikkerhet, ble det ikke observert noen alvorlige bivirkninger i VV116-behandlingsgruppen.
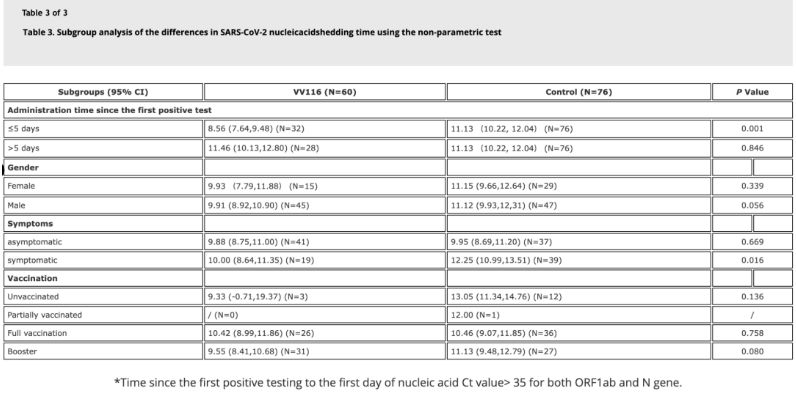
Bildekilde: Referanse 4
Det pågår tre kliniske studier med VV116, hvorav to er fase III-studier på mild til moderat COVID-19 (NCT05242042, NCT05582629). Den andre studien for moderat til alvorlig COVID-19 er en internasjonal multisenter, randomisert, dobbeltblindet fase III klinisk studie (NCT05279235) for å evaluere effekten og sikkerheten til VV116 sammenlignet med standardbehandling. I følge kunngjøringen fra Juniper ble den første pasienten registrert og dosert i mars 2022.

Bildekilde: clinicaltrials.gov
Referanser:
[1]Junshi Biotech: Kunngjøring om hovedendepunktet for fase III-registrert klinisk studie av VV116 versus PAXLOVID for tidlig behandling av mild til moderat COVID-19
[2]https: Yin, Zhiren Fu, Hao Xing, Li Li, Liying Sun, Heyu Huang, Quanbao Zhang, Linlin Xu, Yanting Jin, Rui Chen, Guoyue Lv, Zhijun Zhu, Wenhong Zhang, Zhengxin Wang. (2022) Omicron-infeksjonsprofil og vaksinasjonsstatus blant 1881 levertransplantasjonsmottakere: en multisenter retrospektiv kohort. Emerging Microbes & Infections 11:1, side 2636-2644.
Publisert: 06.01.2023
 Personverninnstillinger
Personverninnstillinger